
The abundance of different miRNAs is usually in ferred from their frequency from the library. To assess the distribution of new miRNAs abundance in drought, salt and pathogen stresses, we normalized the miRNA showed distinct preference of In the past for little RNAs using a distinctive five terminal nucleotide, Also, four OsAGOs which are relevant to AtAGO1 have con served histidine residue from the 798 position that is definitely crit ical for slicer exercise of miRNA.
AGO complex, PF-562271 structure Examination of tiny RNA sequences obtained by immuno precipitation assays with anti AGO1 antibodies uncovered the preferential association of AGO1 with modest RNAs containing five terminal uridine, Related experiments with anti AGO2 and anti AGO4 antibodies showed an enrichment of small RNAs bearing a 5 terminal ad enosine bound to AGO2, and AGO5 related with small RNAs having a 5 terminal cytosine, Primarily based while in the sequence similarity with the sugarcane In the past genes to people of other plant species, it’s pos sible that a similar nucleotide preference may perhaps exist on sugarcane, as well as the benefits in Figure three could indicate the bulk with the new 21 nt microRNA candidates identified on this function are canonical miRNA. Abundance changes of novel sugarcane miRNAs beneath biotic and abiotic stresses Lots of studies have reported the position for miRNA in gene regulation and their involvement in responses to plant stress this kind of as cold, salt, drought and pathogens, reads abundance and used the electronic northern ap proach, The study counts for miRNAs fluctuate very in accordance for the form of pressure. As showed previ ously for soybean, new and known miRNAs have been regulated in water deficit and pathogen assays.
Analyzing the abundance of miRNAs arising from pre cursor class I we uncovered 26 new miRNAs assay kinase inhibitor natural product library specific and 7 miRNAs with abundance higher than 50 normalized reads counts, miRNAs sof miR Seq42, sof miR Seq143, sof miR Seq488, sof miR Seq504, sof miR Seq511 and sof miR Seq656 had been picked for experimental confirmation by stem loop RT PCR process.
These novel miRNAs gave detectable expression amounts in qRT PCR analysis employing controls samples of biotic and abiotic assays, In addition, we observed ex ceptionally high abundance of sof miR Seq513 and sof  miR Seq513 sequences, We confirmed the high expression of this novel miRNA and its miRNA in saline remedy assay sample taken care of for 1 h, The evaluation of miRNAs arising from all precursor classes unveiled differential accumulation of selected new miRNA from the context of the unique anxiety, Only sof miR Seq296 was induced constitutively in all libraries, The biotic pressure library showed larger exclusive expression of new miRNAs, Figure 4A displays the distribution on the 623 novel sugarcane miRNAs uncovered in both remedy or handle samples or in both, Due to the fact the control libraries were constructed with 3 sorts of tissues of various genotypes culti vated in vitro, hydroponic and soil affliction, we ana lyzed the brand new miRNAs distribution in all management conditions, Just one novel miRNA candi date have been shared amongst all handle libraries.
miR Seq513 sequences, We confirmed the high expression of this novel miRNA and its miRNA in saline remedy assay sample taken care of for 1 h, The evaluation of miRNAs arising from all precursor classes unveiled differential accumulation of selected new miRNA from the context of the unique anxiety, Only sof miR Seq296 was induced constitutively in all libraries, The biotic pressure library showed larger exclusive expression of new miRNAs, Figure 4A displays the distribution on the 623 novel sugarcane miRNAs uncovered in both remedy or handle samples or in both, Due to the fact the control libraries were constructed with 3 sorts of tissues of various genotypes culti vated in vitro, hydroponic and soil affliction, we ana lyzed the brand new miRNAs distribution in all management conditions, Just one novel miRNA candi date have been shared amongst all handle libraries.
The abundance of various miRNAs could be in ferred from their fre
Reply
 composed of DNA transposons.
composed of DNA transposons.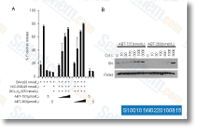 Based on flow cytometry phenotypes without reagent remedy, the 37 mutants can be divided into four groups which have been designated as 2C, 1C, W4C and S4C, respectively. Repre sentative cytometry data of every group are shown in Figure 2A. 2C stands for 2C DNA content. Members of this group, 16 deletions in total, exhibited DNA material peaks at 2C with no reagent therapy, precisely the same as WT cells. Yet, peaks moved towards 1C upon DNA harm triggered by HU or MMS, suggesting that these deletions can cause replication arrest in response to injury. The concentra tffected the outcomes of our display.
Based on flow cytometry phenotypes without reagent remedy, the 37 mutants can be divided into four groups which have been designated as 2C, 1C, W4C and S4C, respectively. Repre sentative cytometry data of every group are shown in Figure 2A. 2C stands for 2C DNA content. Members of this group, 16 deletions in total, exhibited DNA material peaks at 2C with no reagent therapy, precisely the same as WT cells. Yet, peaks moved towards 1C upon DNA harm triggered by HU or MMS, suggesting that these deletions can cause replication arrest in response to injury. The concentra tffected the outcomes of our display. coastal surroundings where the organisms are subject to higher UVB levels that triggers truly serious damages to DNA. The means to resist to UV exposure influences the vertical distribution of seaweeds, and L. dendroidea normally grows while in the decrease midlittoral zone the place UV damage restore may be essential. The identical set of expressed sequences appropriate while in the transcriptome of L. dendroidea are among the most represented in the EST databases of Gracilaria gracilis, G. changii, G. tenuistipitata, Porphyra yezoensis, P. haitanensis, Eucheuma denticulatum, Furcellaria lumbricalis, and Kappaphycus alvarezii, possibly indicating a ge neral pattern of expression in red seaweeds.
coastal surroundings where the organisms are subject to higher UVB levels that triggers truly serious damages to DNA. The means to resist to UV exposure influences the vertical distribution of seaweeds, and L. dendroidea normally grows while in the decrease midlittoral zone the place UV damage restore may be essential. The identical set of expressed sequences appropriate while in the transcriptome of L. dendroidea are among the most represented in the EST databases of Gracilaria gracilis, G. changii, G. tenuistipitata, Porphyra yezoensis, P. haitanensis, Eucheuma denticulatum, Furcellaria lumbricalis, and Kappaphycus alvarezii, possibly indicating a ge neral pattern of expression in red seaweeds. and paraffin sections have been carried out in this examine to survey the microsporogenesis of QS, and it was observed that abnormal tetrads produced in the tetrad stage and subsequently the microsporocyte underwent abnor mal meiosis.
and paraffin sections have been carried out in this examine to survey the microsporogenesis of QS, and it was observed that abnormal tetrads produced in the tetrad stage and subsequently the microsporocyte underwent abnor mal meiosis.